Heterogeneous catalysts are pivotal for the chemical industry and new energy electric conversion technologies, playing a crucial role in advancing low-carbon and green development goals in related fields. Traditional catalyst development models rely heavily on extensive experimental trial-and-error and high-cost theoretical calculations, resulting in long cycles, high costs, and low efficiency. In recent years, a new paradigm of "intelligent R&D" that integrates artificial intelligence (AI) and high-throughput computing has provided an efficient and feasible new path for the rational design and efficient discovery of new catalytic materials from the atomic scale. Associate Professor Wang Xiaonan's research team, affiliated with the Industrial Deep Decarbonization Research Center of the Tsinghua University Institute for Carbon Neutrality and the Department of Chemical Engineering at Tsinghua University, has long been dedicated to this interdisciplinary field. Recently, the team has systematically developed a multi-scale AI prediction framework spanning key stages such as catalyst surface design, catalytic activity and selectivity screening, and reaction kinetics optimization, providing support for the rational design and intelligent discovery of catalysts.
Firstly, addressing the fundamental question of whether a catalytic material surface is stable and can be synthesized, the team created the "SurFF" foundational model for surface properties. Using an active learning strategy, they constructed a large-scale database covering tens of thousands of alloy surfaces to accurately predict the surface energy and equilibrium morphology (Wulff construction) of crystalline materials. While maintaining comparable accuracy, SurFF improves computational efficiency by over five orders of magnitude compared to traditional Density Functional Theory (DFT) calculations. It also achieves, for the first time, high-throughput screening of the synthesizability and surface exposure of thousands of crystalline materials. Simultaneously, the team utilized large language models to mine vast literature data (XRD/TEM facet index information from over 10,000 catalysis publications). Combined with original experimental data, they validated the model's accuracy in real catalytic systems. This work provides robust support for the rational design of catalysts and the development of next-generation self-driven material research cycles. Related results were published in Nature Computational Science on September 9 under the title "SurFF: A foundation model for surface exposure and morphology across intermetallic crystals."
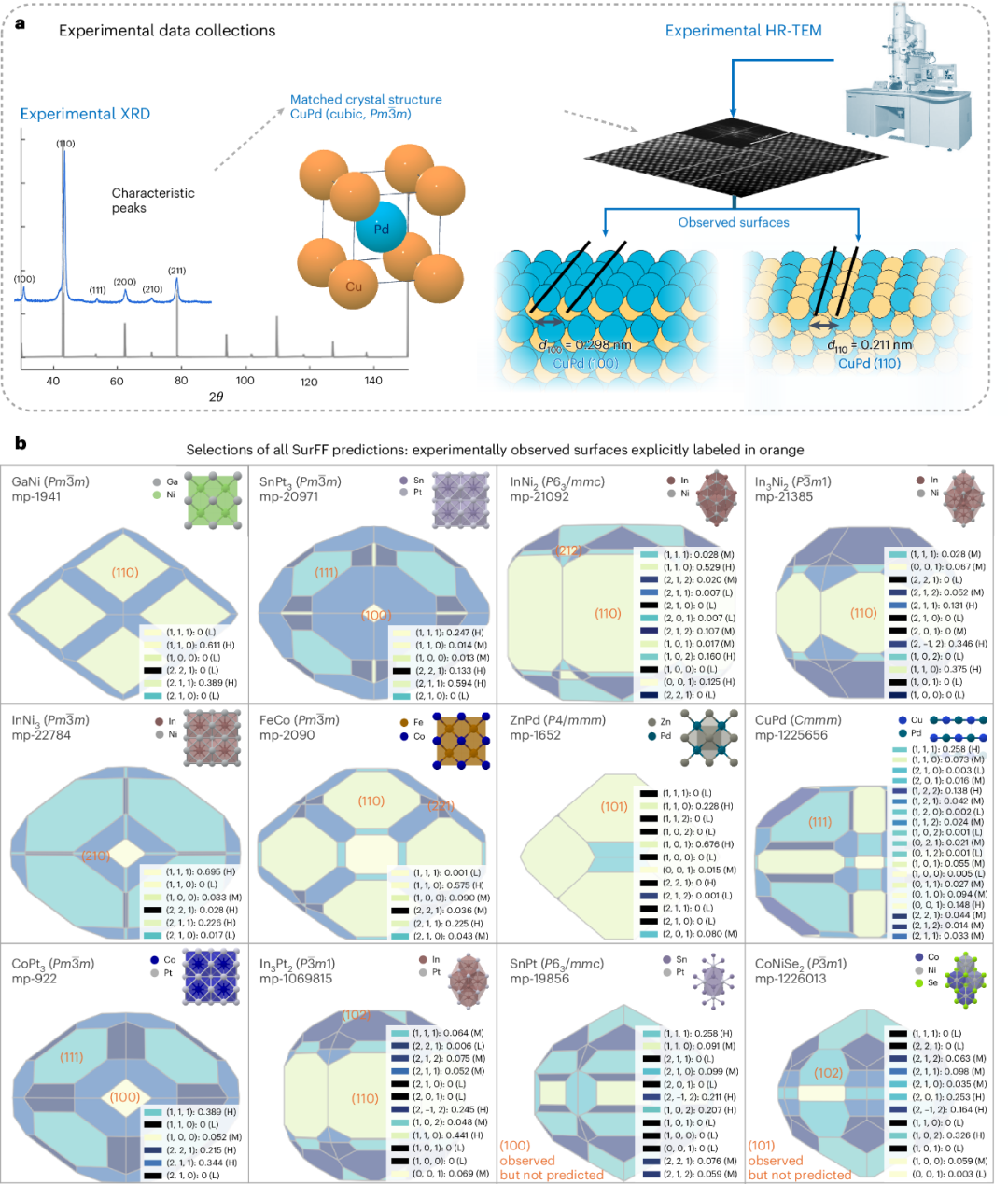
Figure 1. Experimental-level prediction of surface synthesis and exposure using the SurFF foundation model.
Furthermore, the team focused on the significant technical challenge of screening catalysts for activity and selectivity in the electrochemical reduction of CO₂ to methanol. They proposed an AI-driven, high-throughput catalyst screening framework. Based on a pre-trained atomistic foundation model and combined with an active learning strategy, this method can rapidly fine-tune high-precision prediction models using only a small amount of DFT calculation data. This approach improves the screening efficiency for catalyst adsorption properties by over a thousandfold, successfully identifying a series of novel single-atom catalysts with comprehensive performance surpassing existing benchmarks from over 3,000 candidate materials. Related results were published in the journal Engineering (English edition), published by the Chinese Academy of Engineering, on August 5, under the title "Theoretical High-Throughput Screening of Single-Atom CO2 Electroreduction Catalysts to Methanol Using Active Learning."
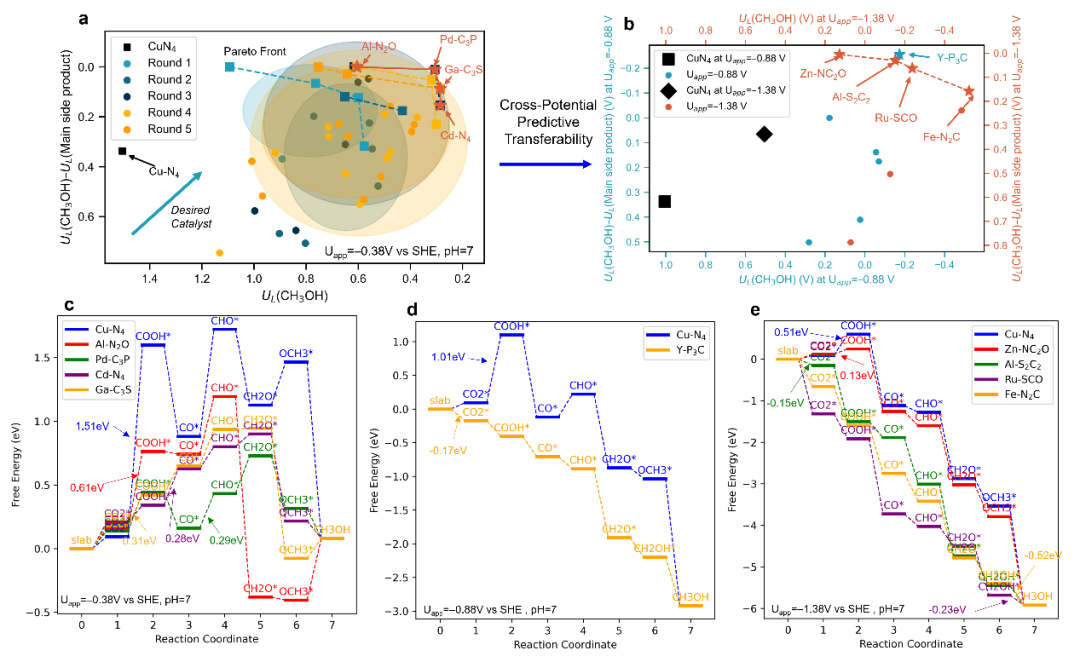
Figure 2. Rational catalyst screening combining active learning and atomistic foundation models.
Going a step further, the team developed a transition state intelligent screening framework named CaTS, tackling the most difficult and time-consuming aspect of catalyst design: reaction kinetics optimization. The calculation of transition states has long been key to predicting reaction rates, but the prohibitive computational cost has been a bottleneck for large-scale screening. The CaTS framework innovatively employs a transfer learning strategy, requiring only several hundred catalytic reaction data points to train a highly accurate machine learning force field. This improves the efficiency of transition state searches by nearly ten thousandfold while maintaining high consistency with DFT in both energy and structural predictions. Related research results were published in ACS Catalysis on August 27, under the title "CaTS: Toward Scalable and Efficient Transition State Screening for Catalyst Discovery."
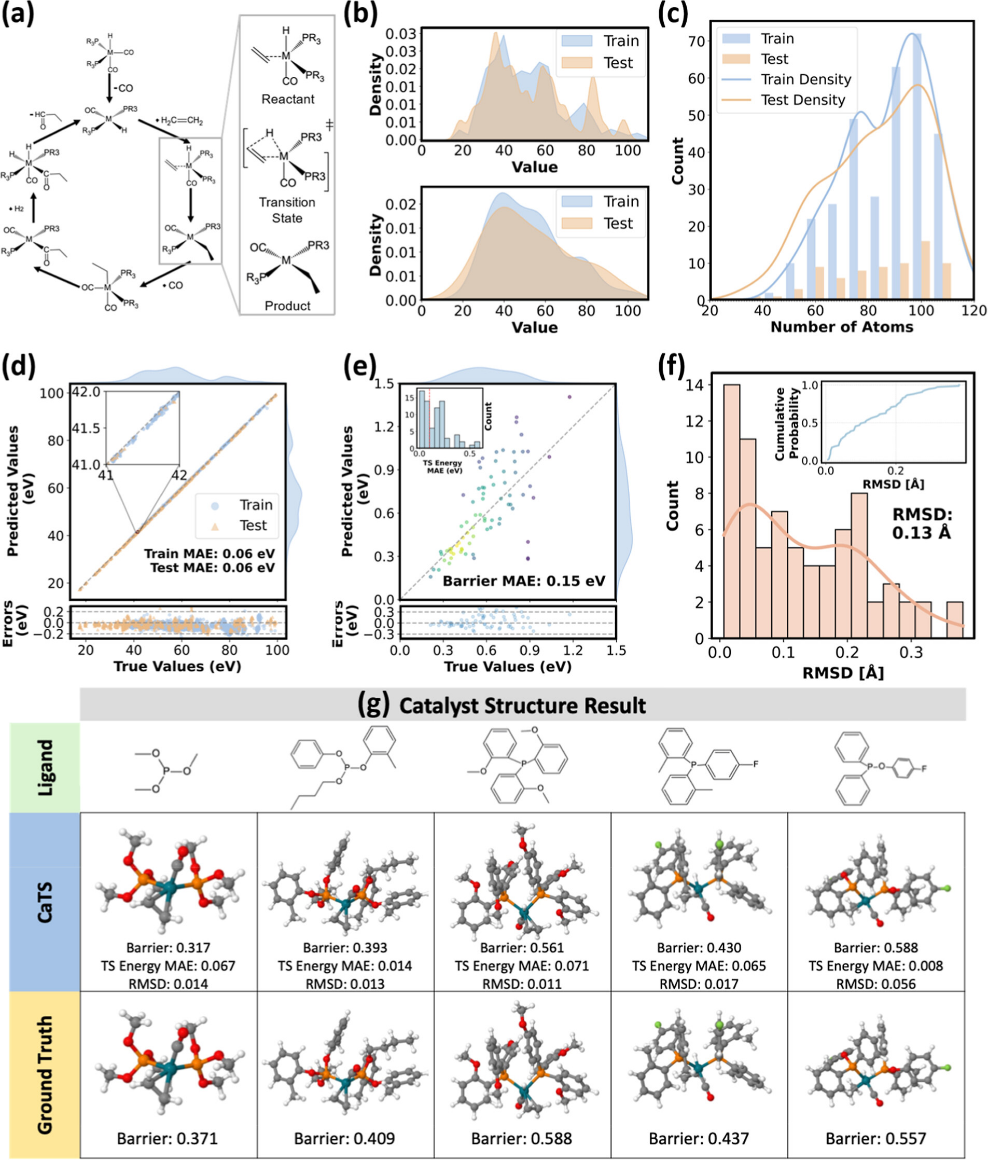
Figure 3. Example of CaTS model transfer and screening application in a homogeneous hydrogenation catalytic system.
These three works address key scientific problems in the rational and intelligent design of catalysts, forming a relatively complete AI-driven multi-scale catalysis research framework that spans from static structure to dynamic reaction. They effectively promote the rapid transition of catalyst R&D from the traditional "trial-and-error" mode to an efficient and precise "intelligent prediction" mode, providing theoretical support and innovative pathway references for the design of various heterogeneous catalysts.
Associate Professor Wang Xiaonan from the Industrial Deep Decarbonisation Research Center of the Institute for Carbon Neutrality and the Department of Chemical Engineering at Tsinghua University is the sole corresponding author for the above series of works. The first authors include Chen Honghao (2022 doctoral student, Tsinghua Chemical Engineering), Li Wentao (2024 doctoral student, Tsinghua Chemical Engineering), and Dr. Yin Jun (recent PhD graduate, National University of Singapore). Other collaborators include Professor Wang Tiefeng and Assistant Researcher Lan Xiaocheng from the Department of Chemical Engineering at Tsinghua University, and Dr. Feng Huasheng from the Sinopec Beijing Research Institute of Chemical Industry. The research was supported by the National Artificial Intelligence Major Project, the Central Universities Young Faculty Research and Innovation Capability Support Project (U40), Tsinghua University Independent Research Project, the Carbon Neutrality and Energy System Transformation (CNEST) Program and the Young Beijing Scholar Program, among others.
Links to the Papers:
https://www.nature.com/articles/s43588-025-00839-0
https://doi.org/10.1016/j.eng.2025.03.039
https://pubs.acs.org/doi/10.1021/acscatal.5c03945



 Latest recommendations
Latest recommendations