2024年4月8日,清华大学碳中和研究院工业深度减碳研究中心副主任、化学工程系魏飞教授课题组在《自然·通讯》上在线发表“Upgrading CO2 to sustainable aromatics via perovskite-mediated tandem catalysis”,标志课题组在芳烃(可再生航煤)合成上取得进展。

研究背景
二氧化碳 (CO2) 是最著名的温室气体,会导致全球变暖以及一系列气候和环境问题。 为了缓解这种人为气候变化,迫切需要开发二氧化碳排放解决方案。 因此,利用废弃二氧化碳作为原料,利用可再生能源将其转化为增值化学品是非常有前途的,既可以回收二氧化碳,又可以减轻社会对化石资源的依赖。 在各种 CO2 利用策略中,利用可再生氢 (H2) 还原能力的热催化 CO2 转化具有生产长链烷烃和重芳烃等复杂分子的独特能力。 从技术经济的角度来看,芳烃是作为合成航空燃料添加剂和许多精细化工应用中的平台分子的理想目标产品。 因此,可再生能源将二氧化碳定向转化为芳烃,为传统的基于化石资源的化学工艺提供了一种可持续且环保的替代方案,因此可以极大地促进航空运输和化学品制造行业的脱碳。
研究者们已经做出了许多努力来开发高效的金属氧化物/沸石复合催化剂,利用CO/CO2加氢产生的氧化物(例如In2O3、ZnZrOx和ZnAlOx等)上的甲醇,随后将它们在沸石组分上转化为芳烃,例如 如 H-ZSM-5。高芳烃选择性源于空间上分离的活性域和串联的过程;当 CO2 和 H2 作为氢化域在氧化物上被激活时,主要生成氢化 C1 物质,而沸石域将这些物质捕获在其有限的酸性孔中,并催化随后向芳烃的复杂转化。通过调节沸石的结构和酸性可以轻松调节选择性。然而,此类催化过程中典型的 CO2 转化率并不令人满意(约 10-30%),这主要受到串联过程中常用的锌/尖晶石基氧化物的缓慢加氢反应的限制。此外,Zn/In物质从金属氧化物迁移到沸石通常会导致催化剂寿命缩短。这些挑战凸显了对创新方法的迫切需求,这些方法不仅要提高二氧化碳转化效率,还要解决催化系统的稳定性和寿命问题。
相反,铁基催化剂已被广泛报道,对于 CO/CO2 加氢生成芳烃具有优异的活性;但一般来说,与 H-ZSM-5 结合时也会出现不理想的选择性。该过程也称为强化费托路线。更具体地说,在铁基氧化物域上生产轻质烯烃(C2-4=),选择性范围为~40-65%。此类中间体扩散到沸石中,开启C-C偶联和环化反应,生成芳烃。然而,这种复合路线包含一个复杂的反应网络:CO2氢化和不受控制的C-C偶联发生在氧化铁上,而进一步的C-C偶联发生在沸石内部。这种复杂性常常导致氧化物和沸石之间的 C-C 偶联速率不匹配,以及沸石通道内烯烃的异构化副反应。因此,在复合催化 CO2 串联转化中,Fe 氧化物对烃类中芳烃的选择性 ( < 70%)很少达到令人满意的水平。虽然极其精确的控制策略(例如引入适量的碱金属)(如 Sun 等人所示)已被证明可以提高选择性,但它们也带来了新的挑战:碱金属往往会迁移到沸石中,随着时间的推移会降低催化性能。为了解决这些问题,将串联系统中的 CO2 加氢域与 C-C 耦合域分离可能是铁基催化剂的另一个潜在策略。换句话说,通过保持强劲的加氢活性,同时调节氧化铁侧的 C-C 偶联倾向,可能为 CO2 高效定向串联转化为芳烃提供巨大潜力。
图文介绍
钙钛矿氧化物的通式为 ABO3,具有共享角的 [BO6] 八面体骨架,其间隙中填充有大的 A 阳离子。在这种结构中,Fe更喜欢占据B位,从而能够在铁基钙钛矿中构建共享角的[FeO6]骨架,这与一般Fe氧化物中的共享面或边的骨架有很大不同。例如 Fe2O3、Fe3O4)和 AFe2O4 型尖晶石(图 1a-d)。这种拓扑差异导致不同的 Fe-Fe 距离,如果氧化物要渗碳,预计会影响 Fe 的迁移(图 1e)。在各种Fe氧化物中,Fe2O3具有最紧密的[FeO6]连接,因此Fe-Fe距离最短,为2.92 Å;AFe2O4 型尖晶石或反尖晶石 Fe3O4 主要含有共边 [FeO6] 八面体,显示中间 Fe-Fe 距离在 2.94-3.10 Å 范围内;Fe 基钙钛矿通常具有更长的 Fe-Fe 距离,> 3.7 Å。在从 Fe 氧化物到 Fe 碳化物的相变过程中,Fe-Fe 距离必须减小到 2.6-2.8 Å 左右。因此,对于铁基钙钛矿来说,相变过程中铁所需的迁移距离显着延长。同时,A位阳离子作为柱桩钉扎在钙钛矿[FeO6]框架中。我们预计钙钛矿的两个结构方面的设计都可以在可能的渗碳过程中抑制铁的迁移,从而实现保留加氢活性,削弱C-C耦连能力。
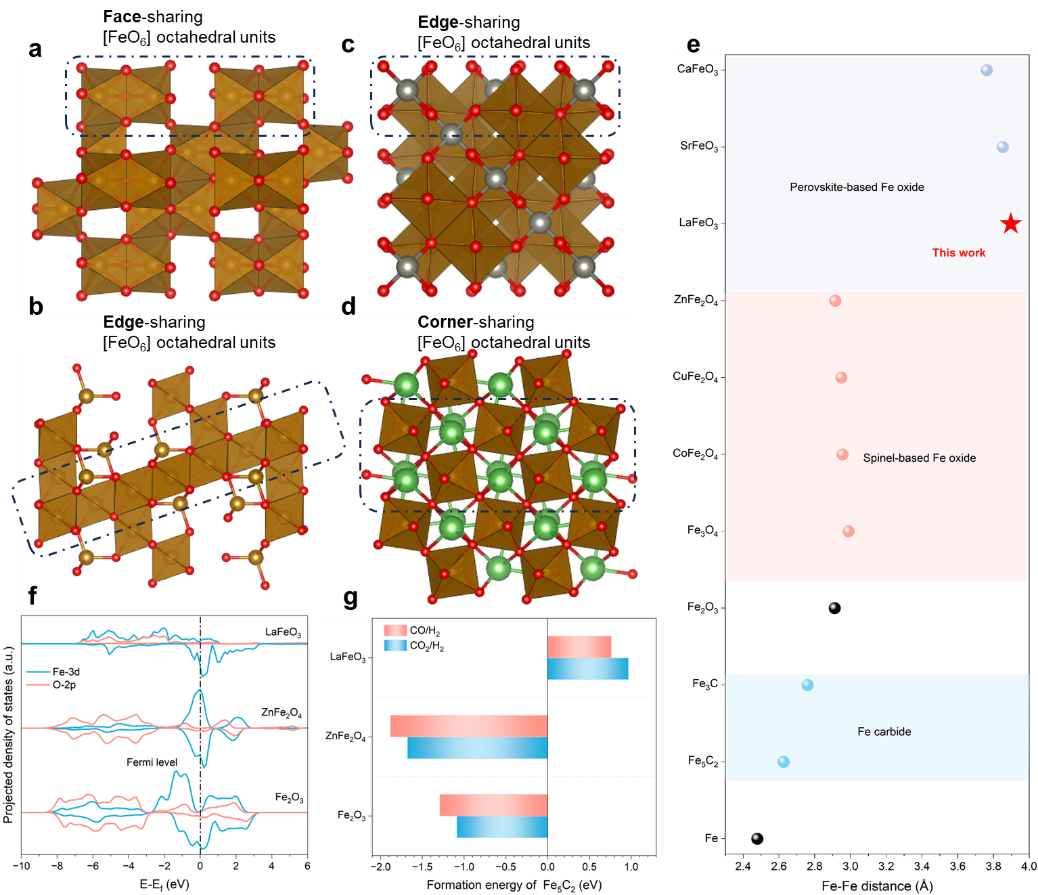
图1. 铁基催化剂中Fe-Fe键长的设计和其C-C耦连,加氢能力的关系
首先对上述三种铁基催化剂在CO2加氢条件下进行了评价。如图 2a 所示,Fe2O3 和 ZnFe2O4 主要产生 C2-4 和 C5+ 范围内的烃(总选择性 > 87%)。这种碳氢化合物分布与 ASF 模型一致,对应于链增长概率 (α) 超过 0.7。相比之下,LaFeO3 产生甲烷 (CH4) 作为主要烃产物 (92.5%),α 大幅降低至 0.09。由于使用后的 Fe2O3 和 ZnFe2O4 氧化物会发生显着的相变,形成 Fe 碳化物,这已被证明是费托合成 (FTS) 中 C-C 偶联的优异催化剂,而使用后的 LaFeO3 则没有。
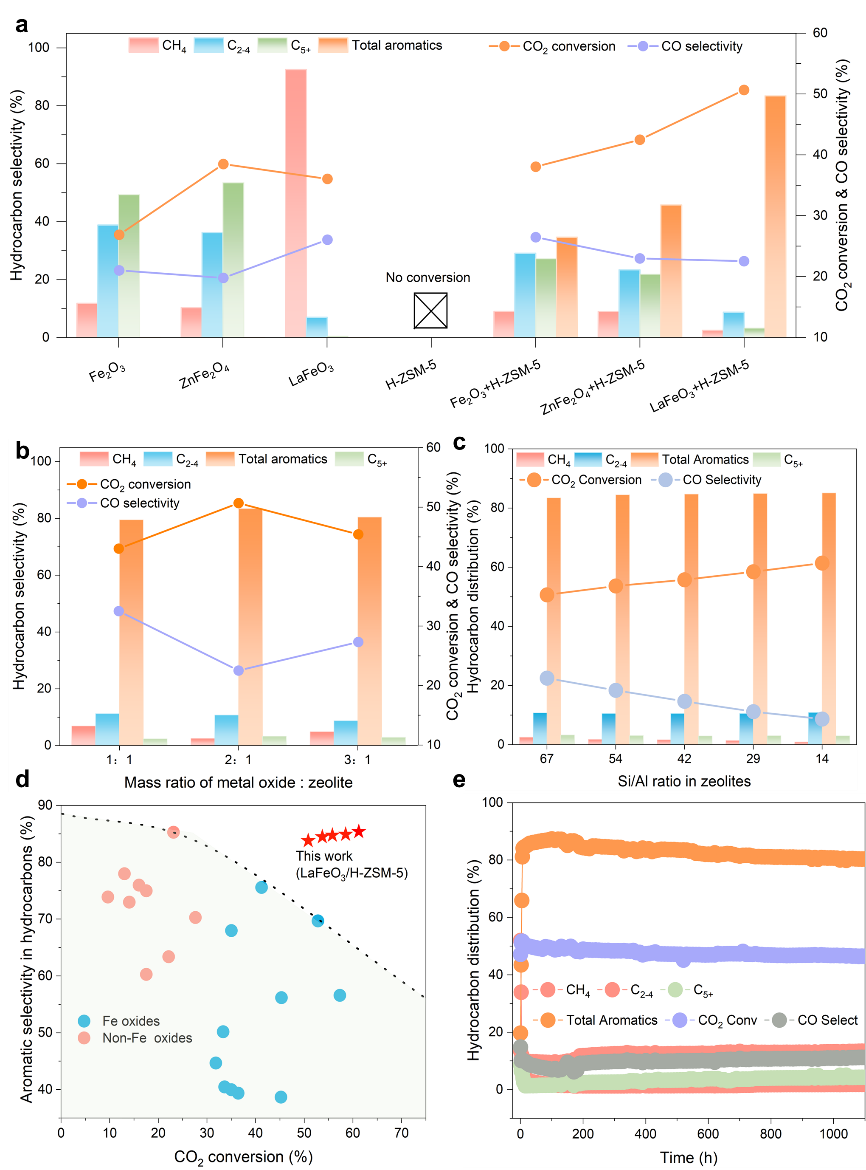
图2. CO2在铁基催化上的催化性能
与 H-ZSM-5(一种硅铝酸盐沸石,能够催化 C-C 偶联和烃异构化,但无法单独催化 CO2 加氢)结合后,观察到产物谱发生显着变化(图 2a)。Fe2O3/H-ZSM-5 和 ZnFe2O4/H-ZSM-5 均表现出 CO2 转化率(38.1% 和 42.5%)和芳烃选择性(34.6% 和 45.8%)的提高,但不如 LaFeO3/ H-ZSM-5(图2a)。C2–4 和 C5+ 烷烃是两个复合系统中的主要碳氢化合物副产品。原位衍生的 Fe 碳化物和 H-ZSM-5 上的两种 C-C 偶联途径之间的竞争分别解释了多碳产物的广泛分布。而LaFeO3/H-ZSM-5在所有碳氢化合物中的总芳烃选择性达到83.8%,而三甲苯和四甲苯是航空燃料中理想的高热值添加剂,占总芳烃选择性的近80% 芳烃(图2a)。此外,CO2转化率从36.1%上升至50.8%。这意味着 H-ZSM-5 可以在原始 LaFeO3 上沿着缓慢的 CO2 氢化途径有效利用某些中间体。这表明此类中间体在 H-ZSM-5 酸性孔中的芳构化可能比氧化物表面上向 CH4 的深度氢化更快。进一步研究了LaFeO3:H-ZSM-5质量比的影响(图2b)。采用 2:1 的适中质量比可获得最佳的 CO2 转化率和芳烃选择性。增加或减少质量比都会导致不必要的催化性能恶化,这归因于互补氧化物和沸石域上的不平衡氢化和 C-C 偶联速率。
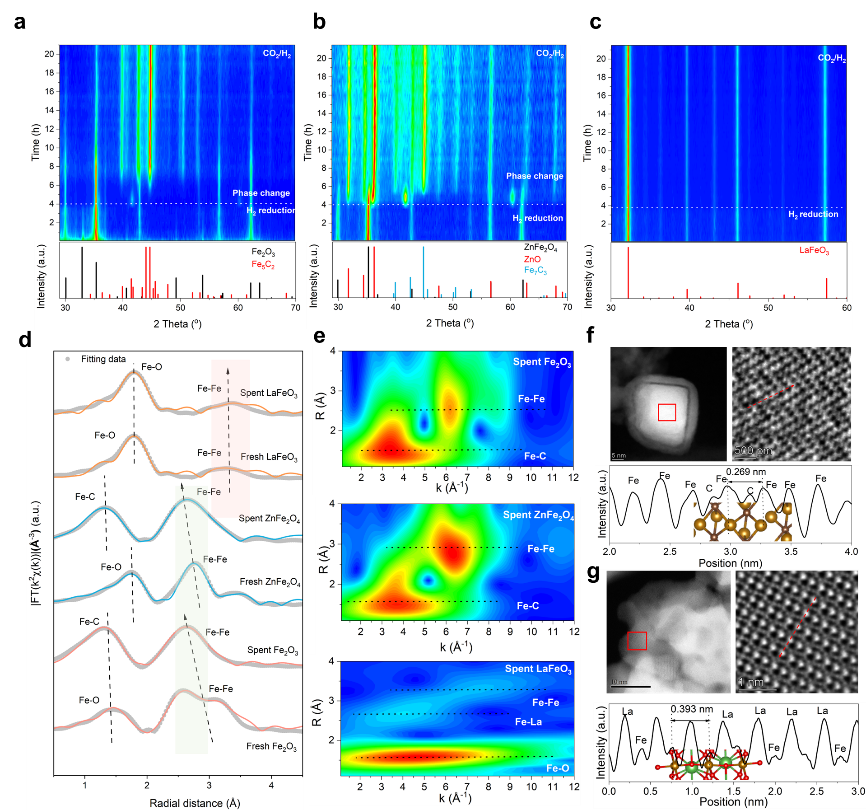
图3. 不同Fe催化剂在反应气氛下的结构演变
LaFeO3 钙钛矿的高芳族选择性和优异的催化稳定性与 LaFeO3 钙钛矿可能成为一种与普通铁氧化物和尖晶石铁氧化物特别不同的抗渗碳催化剂的预期相一致。为了进一步获得反应气氛下催化剂演化的实验见解,进行了原位和异位组合光谱和微观表征。原位 X 射线衍射 (XRD) 首先表明,Fe2O3 和 ZnFe2O4 在初始 H2 还原时可以保持其原始块状结构,但一旦将含碳气体 (CO2) 引入其中,则分别逐渐转变为 Fe5C2 以及 ZnO 和 Fe7C3 的混合物 现场观察室模拟反应气氛(图3a-b)。值得注意的是,LaFeO3 的钙钛矿结构得到了很好的保留,在 H2 或 CO2/H2 气氛下没有产生新的晶相(图 3c)。
进一步采用异位同步加速器 X 射线表征来研究废氧化铁催化剂的化学状态和精细局部结构。归一化的 Fe K 边 X 射线吸收近边结构揭示了反应 24 小时后 Fe2O3 和 ZnFe2O4 中 Fe 的氧化态显着降低,与原位 XPS 观察结果一致。在 FT-EXAFS 中,Fe2O3 和 ZnFe2O4 反应 24 小时后,分配给 Fe-Fe 配位的第二壳层峰转移到距离较短的位置(图 3d)。使用后的Fe2O3和ZnFe2O4中的Fe-Fe配位数均增加至7以上,表明形成了具有更多团聚Fe的Fe碳化物。与 Fe2O3 和 ZnFe2O4 不同,LaFeO3 在相同处理后表现出 Fe 氧化态、Fe-Fe 距离(最初比 Fe2O3 和 ZnFe2O4 长)和 Fe-Fe 配位数略有变化。WT-EXAFS 提供了有关催化剂演变的更多见解(图 3e)。使用后的LaFeO3 中的Fe-Fe 散射强度低于使用后的Fe2O3 和尖晶石ZnFe2O4,表明Fe-Fe 相互作用较弱,从而抑制了Fe 迁移和聚集。
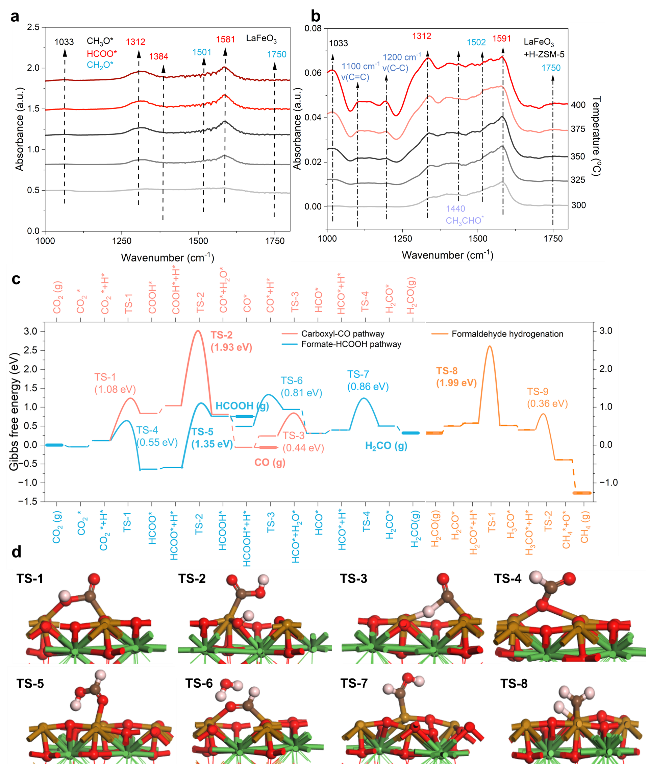
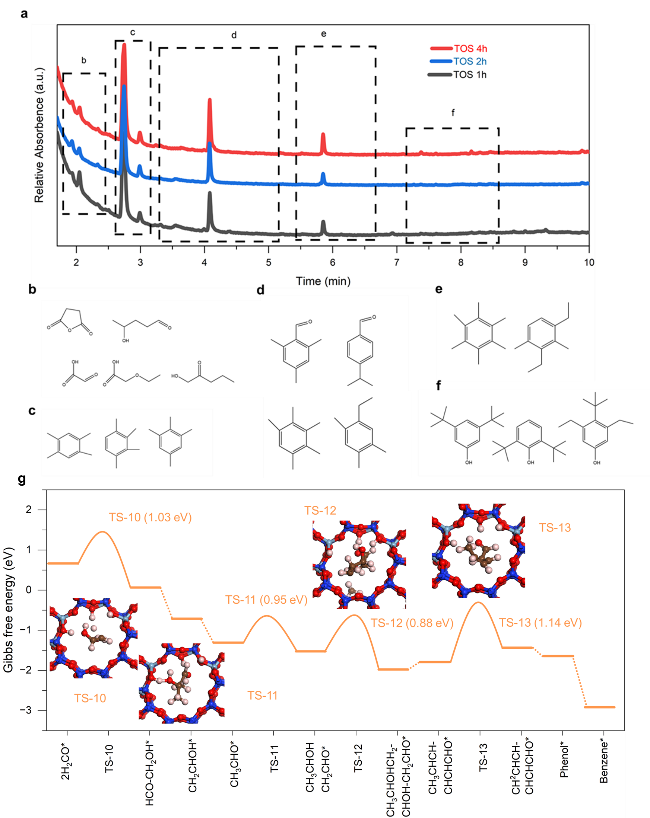
图4. (左) LaFeO3 上 CO2 转化的机理研究 (右)中间体迁移到分子筛的后续反应
反应热力学和动力学使一些实验观察结果合理化。首先,通过原位谱学探测,主要的甲酸途径会产生大量含氧物质。其次,HCOOH (0.75 eV) 和 H2CO (0.31 eV) 的弱吸附,以及 H2CO 进一步氢化的高势垒 (1.99 eV,TS-8),决定了纯 LaFeO3 和 LaFeO3/H-ZSM-5 之间 CO2 转化的差异。在纯 LaFeO3 上,这些物质的容易解吸会降低其表面覆盖率,或者等效地,如果我们假设从 HCOOH 或 H2CO 气体开始,则增加后续氢化的自由能损失,这两种气体都在氢化反应过程中在纯 LaFeO3 上通过实验检测到。而对于 LaFeO3/H-ZSM-5,H-ZSM-5 酸性孔中解吸的 HCOOH 和/或 H2CO 的转化在热力学上非常有利于芳烃),并且芳构化的障碍已被证明相当低。因此,在整个系统中加入H-ZSM-5后,实现了CO2转化率的提高。第三,由于HCO*分解(后向;TS-3)的势垒比HCO*加氢(前向,TS-7)更低,因此通过间接途径(CO2→HCOO*→HCOOH→HCO*)形成CO → CO) 比直接的羧基途径更可行。这种理论见解可能会启发未来调节表面甲酰基的吸附和转化以实现低CO选择性的努力。
为了证实 HCOOH/H2CO 作为串联催化中的关键中间体,我们进一步探索了 H-ZSM-5 (Si/Al=15)内的醇醛芳香反应。执行了由先前方法开发的一个实验程序来检测不同运行时间(TOS)期间沸石内的物质。可以发现一些羟醛缩合物、芳香醛和酚类芳烃以及多甲苯(图5a-f)。羟醛缩合物从1h到4h的减少表明羟醛反应在诱导初期发挥了重要作用。此外,有证据表明,在诱导1h和2h的TOS中,芳香醛、酚类也表现出相当数量,这表明羟醛缩合物转化为芳香醛和一些酚类含氧化合物的可行性。此外,还通过H-ZSM-5内部的DFT计算考虑了详细的反应路线。我们假设 CH2O 物质快速扩散到 H-ZSM-5 中,引发一系列向醛醇缩合物的转化。具体来说,两个 H2CO 分子最初吸附在 Al-OH 位点附近,引发第一次 C-C 偶联。该过程导致 CH3CHO 物质的形成(也在图 4b 中观察到),然后进入羟醛反应,促进进一步的 C-C 偶联,直到形成长链六碳含氧化合物。随后,它们经历环化和芳构化,这一过程受到 H-ZSM-5 的限制和酸性特性的影响,最终导致苯的形成。从热力学角度来看,H-ZSM-5 内的能量分布明显低于 LaFeO3 表面上 C1 物质的能量分布。这一观察结果表明,沸石有效地将反应途径从 CO2-H2CO-CH4 改变为更理想的 CO2-H2CO-芳烃过程。这种计算见解与我们的实验结果一致,为观察到的转化途径提供了强有力的理论依据,并强调了 HCOOH/H2CO 作为中间体在这种复杂的串联催化过程中的关键作用。
本文共同第一作者为清华大学化工系博士生田果、李正稳,清华大学碳中和研究院工业深度减碳研究中心、化工系魏飞教授、张晨曦副研究员、陈晓副研究员,电子科技大学基础与前沿研究院研究员彭翃杰是该文的通讯作者。上述研究工作得到国家重点研发计划、国家自然科学基金的支持。
文献信息
Upgrading CO2 to sustainable aromatics via perovskite-mediated tandem catalysis
https://www.nature.com/articles/s41467-024-47270-z.pdf



 最新推荐
最新推荐